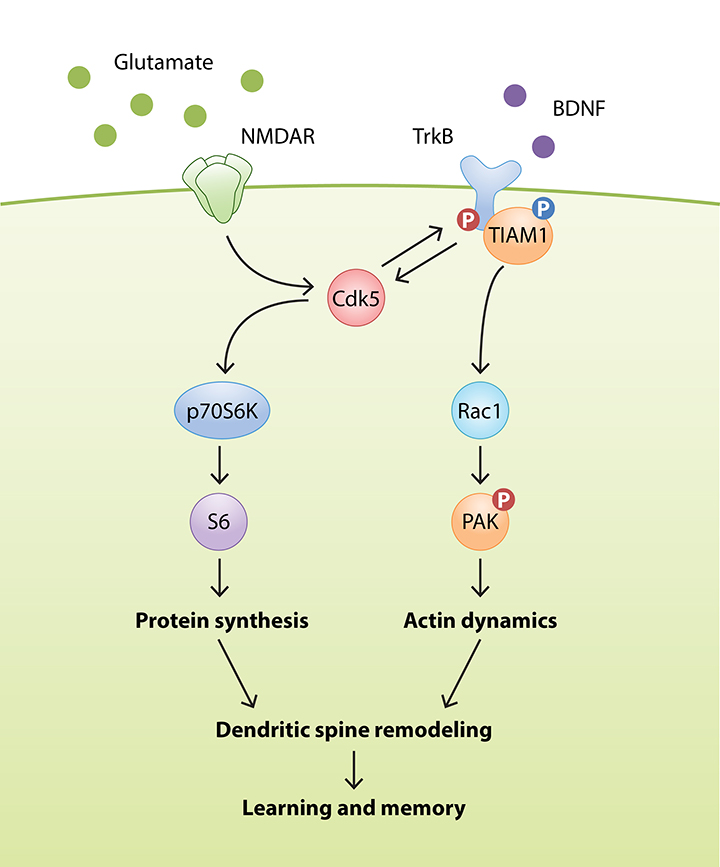
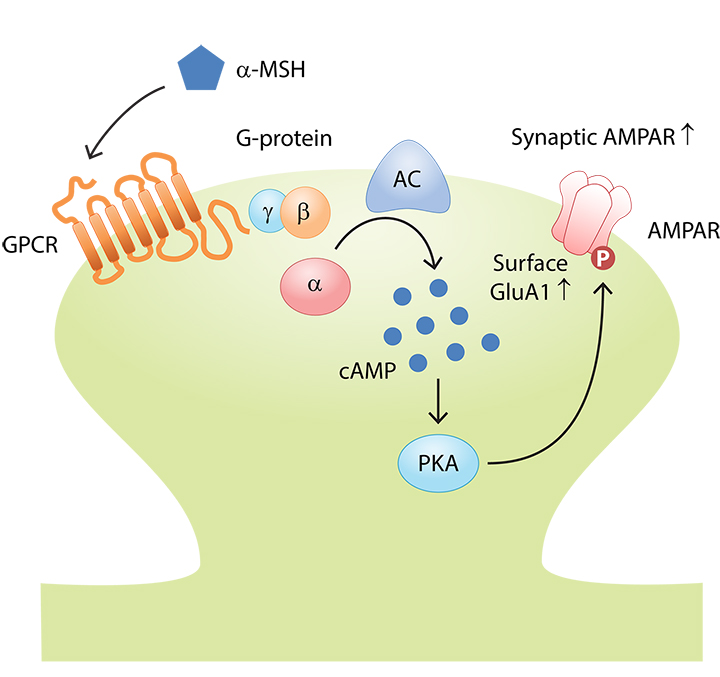
Experience-dependent changes in synaptic transmission, i.e., synaptic plasticity, form the cellular basis of cognitive functions such as learning and memory. Synaptic plasticity impairment can be devastating and is believed to underlie various psychiatric disorders and neurodegenerative diseases. Using a combination of molecular, cellular, electrophysiological, and behavioral approaches, our laboratory aims to elucidate the signaling pathways that regulate synaptic function in the adult brain under normal and diseased conditions. In particular, we aim to determine how protein phosphorylation relays extracellular signals to modulate synapse formation, maturation, and plasticity.